
Compartir:
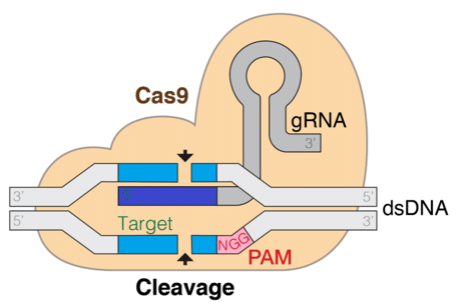
La tecnología de repetición palindrómica interespaciada de manera regular y agrupada (CRISPR) promete revolucionar las tecnologías de edición genética, lo que es transformador para la comprensión y tratamiento de enfermedades. Esta técnica se basa en un mecanismo natural encontrado en bacterias que permite que una proteína, acoplada a una cadena de ARN guía (gRNA), localice y haga cortes específicos en el genoma objetivo. Poder predecir computacionalmente la eficiencia y especificidad del gRNA es fundamental para el éxito de la edición genética.
El ARN, transcrito a partir de secuencias de ADN, es un tipo importante de secuencia biológica de ribonucleótidos (A, U, G, C), que se pliega en una estructura tridimensional. Aprovechando los avances recientes en modelos de lenguaje de gran escala (LLMs), se pueden resolver una variedad de tareas en biología computacional ajustando modelos biológicos de LLM preentrenados en miles de millones de secuencias biológicas conocidas. Las tareas descendentes en ARNs están relativamente poco estudiadas.
En esta investigación, adoptamos un LLM genómico preentrenado para la predicción de eficiencia del gRNA. La idea es tratar un gRNA diseñado por computadora como una oración y ajustar el LLM para realizar tareas de regresión a nivel de oración, análogas al análisis de sentimientos. Utilizamos métodos de ajuste fino eficientes en parámetros para reducir el número de parámetros y el uso de GPU para esta tarea.
Los modelos de lenguaje grande (LLMs) han ganado mucho interés por su capacidad para codificar la sintaxis y la semántica de los lenguajes naturales. La arquitectura neuronal detrás de los LLMs son transformadores, que constan de bloques codificadores-decodificadores basados en atención que generan una representación interna de los datos de los que están entrenados (codificador) y son capaces de generar secuencias en el mismo espacio latente que se asemejan a los datos originales (decodificador). Debido a su éxito en el lenguaje natural, trabajos recientes han explorado el uso de LLMs para información de biología molecular, que es secuencial por naturaleza.
DNABERT es un modelo transformador preentrenado con datos de secuencias de ADN humano sin solapamiento. La columna vertebral es una arquitectura BERT compuesta por 12 capas de codificación. Los autores de este modelo informan que DNABERT es capaz de capturar una buena representación de características del genoma humano, lo que permite un rendimiento de vanguardia en tareas descendentes como la predicción de promotores y la identificación de sitios de empalme y unión. Decidimos usar este modelo como base para nuestros experimentos.
A pesar del éxito y la adopción popular de los LLMs, ajustar estos modelos puede ser difícil debido al número de parámetros y la computación necesaria. Por esta razón, se han desarrollado métodos de Ajuste fino eficiente en parámetros (PEFT). En esta investigación, utilizamos uno de estos métodos, llamado LoRA (Adaptación de Baja Rango). Presentamos el método en las siguientes secciones.
El objetivo de esta solución es ajustar el modelo base DNABERT para predecir la eficiencia de actividad de diferentes candidatos de gRNA. Por lo tanto, nuestra solución primero toma datos de gRNA y los procesa, como se describe más adelante. Luego, usamos un notebook de Amazon SageMaker y la biblioteca PEFT de Hugging Face para ajustar el modelo DNABERT con los datos de ARN procesados. La etiqueta que queremos predecir es la puntuación de eficiencia tal como se calculó en condiciones experimentales probando con las secuencias de ARN reales en cultivos celulares. Esas puntuaciones describen un equilibrio entre ser capaz de editar el genoma y no dañar el ADN que no se tenía como objetivo.
Para esta solución, se necesita acceso a los siguientes elementos:
– Una instancia de notebook de SageMaker (entrenamos el modelo en una instancia ml.g4dn.8xlarge con una sola GPU NVIDIA T4)
– transformers-4.34.1
– peft-0.5.0
– DNABERT 6
Usamos los datos de gRNA liberados por investigadores en un artículo sobre predicción de gRNA utilizando aprendizaje profundo. Este conjunto de datos contiene puntuaciones de eficiencia calculadas para diferentes gRNAs.
Para entrenar el modelo, se necesita una secuencia de gRNA de 30-mers y una puntuación de eficiencia. Un k-mer es una secuencia contigua de k bases nucleotídicas extraída de una secuencia más larga de ADN o ARN. Por ejemplo, si se tiene la secuencia de ADN “ATCGATCG” y se elige k = 3, los k-mers dentro de esta secuencia serían “ATC,” “TCG,” “CGA,” “GAT,” y “ATC.”
Para codificar la secuencia de gRNA, utilizamos el codificador DNABERT. DNABERT se entrenó previamente con datos genómicos humanos, por lo que es un buen modelo para codificar secuencias de gRNA. DNABERT tokeniza la secuencia de nucleótidos en k-mers superpuestos, y cada k-mer sirve como una palabra en el vocabulario del modelo DNABERT. La secuencia de gRNA se divide en una secuencia de k-mers, y luego cada k-mer se reemplaza por una incrustación para el k-mer en la capa de entrada. De lo contrario, la arquitectura de DNABERT es similar a la de BERT. Después de codificar el gRNA, utilizamos la representación del token [CLS] como la codificación final de la secuencia gRNA. Para predecir la puntuación de eficiencia, utilizamos una capa de regresión adicional. El MSE será el objetivo de entrenamiento.
Ajustar todos los parámetros de un modelo es costoso porque el modelo preentrenado se vuelve mucho más grande. LoRA es una técnica innovadora desarrollada para abordar el desafío de ajustar modelos de lenguaje extremadamente grandes. LoRA ofrece una solución sugiriendo que los pesos del modelo preentrenado permanezcan fijos mientras se introducen capas entrenables (denominadas matrices de descomposición de rango) dentro de cada bloque transformador. Este enfoque reduce significativamente el número de parámetros que necesitan ser entrenados y disminuye los requisitos de memoria de GPU, porque la mayoría de los pesos del modelo no requieren cálculos de gradiente.
Por lo tanto, adoptamos LoRA como un método PEFT en el modelo DNABERT. LoRA está implementado en la biblioteca PEFT de Hugging Face. Cuando se usa PEFT para entrenar un modelo con LoRA, los hiperparámetros del proceso de adaptación de bajo rango y la forma de envolver modelos de transformadores base pueden definirse.
Se utilizaron RMSE, MSE y MAE como métricas de evaluación, y se probó con rangos de 8 y 16. Además, se implementó un método de ajuste fino simple, que simplemente agrega varias capas densas después de las incrustaciones de DNABERT. La siguiente tabla resume los resultados:
| Método | RMSE | MSE | MAE |
|————————|——–|———-|——-|
| LoRA (rango = 8) | 11.933 | 142.397 | 7.014 |
| LoRA (rango = 16) | 13.039 | 170.010 | 7.157 |
| Una capa densa | 15.435 | 238.265 | 9.351 |
| Tres capas densas | 15.435 | 238.241 | 9.505 |
| CRISPRon | 11.788 | 138.971 | 7.134 |
Cuando el rango es 8, se tienen 296,450 parámetros entrenables, que es aproximadamente el 33% de la totalidad. Las métricas de desempeño son «rmse»: 11.933, «mse»: 142.397, «mae»: 7.014.
Cuando el rango es 16, se tienen 591,362 parámetros entrenables, que es aproximadamente el 66% de la totalidad. Las métricas de desempeño son «rmse»: 13.039, «mse»: 170.010, «mae»: 7.157, lo que podría indicar un problema de sobreajuste bajo esta configuración.
Finalmente, comparamos con el método existente CRISPRon, que es un modelo de aprendizaje profundo basado en CNN. Las métricas de desempeño son «rmse»: 11.788, «mse»: 138.971, «mae»: 7.134.
Como se esperaba, LoRA funciona mucho mejor que simplemente agregar algunas capas densas. Aunque el rendimiento de LoRA es un poco peor que el de CRISPRon, con una búsqueda exhaustiva de hiperparámetros, es probable que supere a CRISPRon.
Recomendamos encarecidamente apagar la instancia de SageMaker cuando no esté en uso activo, para evitar costos innecesarios en la computación no utilizada.
En conclusión, mostramos cómo usar métodos PEFT para ajustar modelos de lenguaje de ADN utilizando SageMaker, enfocándonos en predecir la eficiencia de las secuencias de ARN de CRISPR-Cas9 para su impacto en las tecnologías actuales de edición genética. También proporcionamos código que puede ayudar a comenzar con sus aplicaciones de biología en AWS.









